
New Scientist – Mice with a form of dementia have had the condition reversed by a process that involves ‘rebooting’ brain cells otherwise destined to die.
The process that kills the cells could be common to all dementias, so blocking it in the same way might hold promise for Alzheimer’s disease and Parkinson’s disease – although more research is needed to explore this further.
“This is potentially a common pathway in all these diseases,” says Giovanna Mallucci at the University of Leicester, UK. “The key thing is that we’ve moved away from a disease-specific mechanism to a more generic cause of cell death,” she says.
Instead of trying to tackle the prions or plaques – an approach that has so far proved unsuccessful in Alzheimer’s disease – Mallucci’s team tackled another process that goes wrong in the affected brain cells: a complete shutdown of protein production.
Mallucci’s team restarted protein production with a treatment that stripped off the phosphate group. They did this by using a virus to load into the mice’s brains extra amounts of another protein, called GADD34, which snips off the crucial phosphate from the eIF2alpha-P protein.
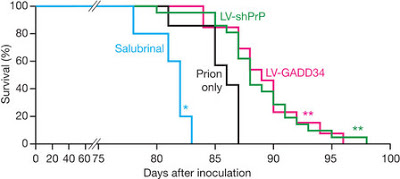
By rebooting protein production, the treatment halted any further degeneration in the mice. Post mortems showed that brain connections lost in the untreated mice remained healthy, and completely normal protein production had resumed in the treated animals, even though the prions continued to accumulate. The treatment slightly extended the lifespan of the treated mice too. On average they died after 90 days. Untreated mice died after 83 days on average.
Nature – Sustained translational repression by eIF2α-P mediates prion neurodegeneration

Reducing eIF2α-P levels in prion-diseased mice significantly increases survival.
The mechanisms leading to neuronal death in neurodegenerative disease are poorly understood. Many of these disorders, including Alzheimer’s, Parkinson’s and prion diseases, are associated with the accumulation of misfolded disease-specific proteins. The unfolded protein response is a protective cellular mechanism triggered by rising levels of misfolded proteins. One arm of this pathway results in the transient shutdown of protein translation, through phosphorylation of the α-subunit of eukaryotic translation initiation factor, eIF2. Activation of the unfolded protein response and/or increased eIF2α-P levels are seen in patients with Alzheimer’s, Parkinson’s and prion diseases but how this links to neurodegeneration is unknown. Here we show that accumulation of prion protein during prion replication causes persistent translational repression of global protein synthesis by eIF2α-P, associated with synaptic failure and neuronal loss in prion-diseased mice. Further, we show that promoting translational recovery in hippocampi of prion-infected mice is neuroprotective. Overexpression of GADD34, a specific eIF2α-P phosphatase, as well as reduction of levels of prion protein by lentivirally mediated RNA interference, reduced eIF2α-P levels. As a result, both approaches restored vital translation rates during prion disease, rescuing synaptic deficits and neuronal loss, thereby significantly increasing survival. In contrast, salubrinal, an inhibitor of eIF2α-P dephosphorylation5, increased eIF2α-P levels, exacerbating neurotoxicity and significantly reducing survival in prion-diseased mice. Given the prevalence of protein misfolding and activation of the unfolded protein response in several neurodegenerative diseases, our results suggest that manipulation of common pathways such as translational control, rather than disease-specific approaches, may lead to new therapies preventing synaptic failure and neuronal loss across the spectrum of these disorders.
If you liked this article, please give it a quick review on ycombinator or StumbleUpon. Thanks

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.