

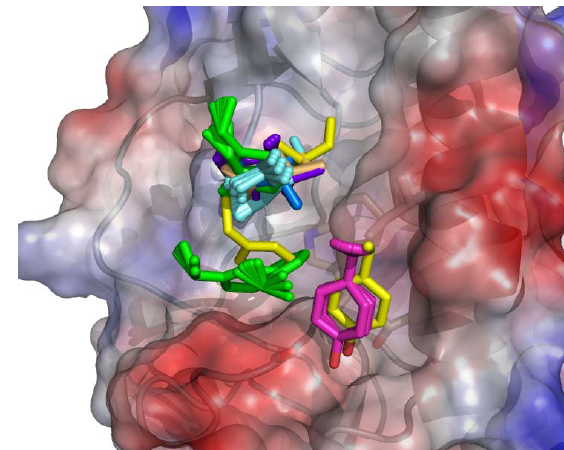
The authors from the Baker lab used RosettaDesign-based protein engineering tools to design proteins with surface structures that bind to a natural protein at a particular location, and with a particular orientation. Finding a protein that binds isn’t too hard — screening and evolutionary methods applied to antibodies (among other proteins) can do this — but achieving high affinity (tight binding) in a specific geometry is new.
They achieved this by designing binders with the correct geometry but mediocre binding, and then using selection (the equivalent of antibody affinity maturation) to refine the interfaces to achieve high affinity. The refinement process retains the initial alignment with good fidelity.
The authors didn’t address the problem of designing building-block interfaces, as an engineer would understand the task: They did something harder. Only side of the interface was designed to bind, while the other was a naturally occurring structure that normally binds nothing. An engineer designing building-block assemblies, by contrast, would design the interface as a unit, not just one side of it. Both sides can be tweaked for the best fit.
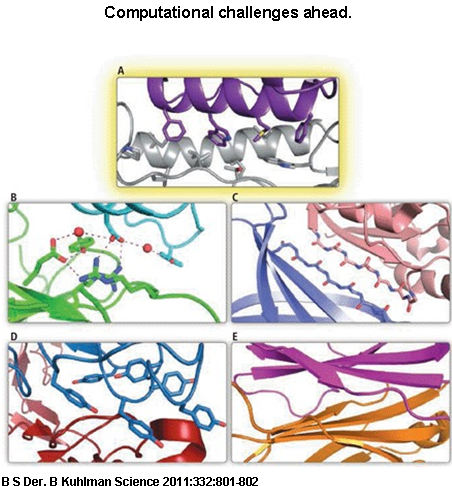
Computational challenges ahead. The diversity of protein interface characteristics observed in nature suggests future challenges for computational design. (A) Fleishman et al. designed a hydrophobic helix (purple) to bind a hydrophobic groove (gray) with unprecedented accuracy in binding location and orientation. (B) The high-affinity interaction between the bacterial proteins barnase and barstar features a sophisticated hydrogen bond network that also includes water molecules. (C) Strand-strand pairings at an interface feature regular repeats of polar atoms. (D) Imitating an antibody interface that features long loops will require precise backbone conformational sampling and scoring methods. Loops provide a rich diversity of backbone conformations, such that binding can occur using only tyrosine and serine side chains (5). (E) The quaternary structure of an antibody is stabilized by a sheet-sheet interface. CREDIT: BRYAN S. DER AND BRIAN KUHLMAN”
Journal Science – From Computational Design to a Protein That Binds
RosettaDesign server for protein design
The RosettaDesign server identifies low energy amino acid sequences for target protein structures (http://rosettadesign.med.unc.edu). The client provides the backbone coordinates of the target structure and specifies which residues to design. The server returns to the client the sequences, coordinates and energies of the designed proteins. The simulations are performed using the design module of the Rosetta program (RosettaDesign). RosettaDesign uses Monte Carlo optimization with simulated annealing to search for amino acids that pack well on the target structure and satisfy hydrogen bonding potential. RosettaDesign has been experimentally validated and has been used previously to stabilize naturally occurring proteins and design a novel protein structure
53 pages of supplemental material
If you liked this article, please give it a quick review on ycombinator or StumbleUpon. Thanks

Brian Wang is a Futurist Thought Leader and a popular Science blogger with 1 million readers per month. His blog Nextbigfuture.com is ranked #1 Science News Blog. It covers many disruptive technology and trends including Space, Robotics, Artificial Intelligence, Medicine, Anti-aging Biotechnology, and Nanotechnology.
Known for identifying cutting edge technologies, he is currently a Co-Founder of a startup and fundraiser for high potential early-stage companies. He is the Head of Research for Allocations for deep technology investments and an Angel Investor at Space Angels.
A frequent speaker at corporations, he has been a TEDx speaker, a Singularity University speaker and guest at numerous interviews for radio and podcasts. He is open to public speaking and advising engagements.